
What is LDS?
Loeys-Dietz syndrome (LDS) is a rare genetic condition that affects the body’s connective tissue. This can alter many parts of the body including the aorta, heart, arteries, skin, and bones. When there is significant enlargement of the artery (an aneurysm), there is a risk of the artery wall tearing (aortic dissection) which can be life-threatening. Many different genetic mutations can cause people to develop LDS. These mutations can occur in any of the following genes: TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, SMAD2.
LDS can either be inherited from a parent with the genetic defect or it may be due to a new genetic change (spontaneous mutation) in an individual. Spontaneous mutation occurs in about 3 out of 4 (75%) people who have LDS.
Doctors classify LDS into different subtypes depending on which gene is affected:
- Loeys-Dietz syndrome 1 caused by changes in the TGFBR1 gene (found in 20-25% of patients)
- Loeys-Dietz syndrome 2 caused by changes in the TGFBR2 gene (found in 55-60% of patients)
- Loeys-Dietz syndrome 3 caused by changes in the SMAD3 gene (found in 5-10% of patients)
- Loeys-Dietz syndrome 4 caused by changes in the TGFB2 gene (found in 5-10% of patients)
- Loeys-Dietz syndrome 5 caused by changes in the TGFB3 gene (found in 1-5% of patients)
- Loeys-Dietz syndrome 6 caused by changes in the SMAD2 gene (found in 1-5% of patients)
There is a lot of overlap in the clinical features of the different LDS subtypes. Since many of these genes were only recently discovered, we are still learning a lot about how they differ. Most of the information about LDS comes from the most common types, LDS1 and LDS2. In the very first cases described, it was common to see very aggressive aortic and arterial disease. More recently, we have found that there is more variation in the severity of the disease. This is because, with more widespread genetic testing and better recognition of the features of the condition, we can identify people with even mild LDS. Also, the prognosis for people with LDS has improved over the years because of better recognition and preventative aortic surgery for enlargement of the aorta and branches.
LDS3 has many of the typical features of LDS, but also has more arthritis of the spine, knees, and ankles. LDS3 is also called Aneurysms-Osteoarthritis syndrome (AOS). LDS4 has some overlapping features of LDS and Marfan syndrome. There is very little information about LDS5 or LDS6.
How common is LDS?
LDS is rare, but the prevalence is unknown. LDS types 1 and 2 are the most common forms.
What are the features of LDS?
There are many features that can be seen in some, but not all, people with LDS. These include:
- Heart and Blood Vessels
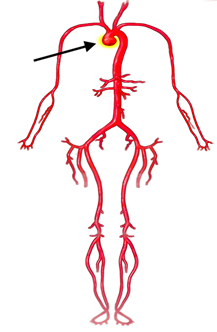
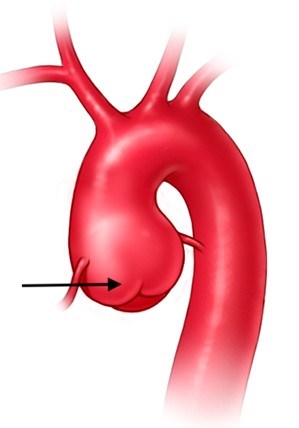
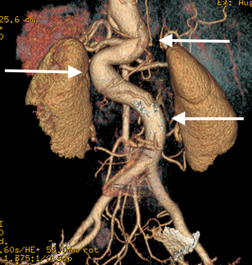
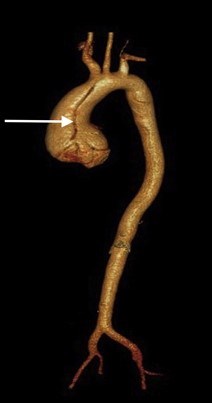
- Dilation or enlargement of aorta and arteries coming from the aorta (aneurysms) (Figures 1-2)
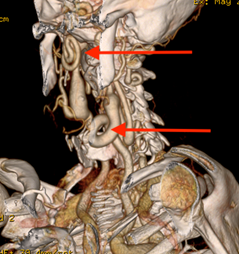
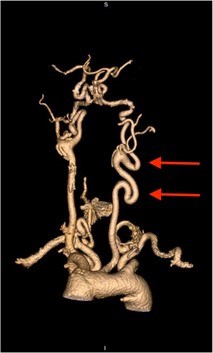
- Abnormal twisting or spiraling of the arteries (arterial tortuosity) (Figures 3-5)
- Changes to the heart structure at birth (congenital heart defects)
- PDA (patent ductus arteriosus), ASD (atrial septal defect), VSD (ventricular septal defect), BAV (bicuspid aortic valve)
- Aortic dissection (a tear or split in the lining of the aortic wall that allows blood to enter the aortic wall) (Figure 6)
- Mitral valve prolapse (floppy mitral valve) which may lead to mitral regurgitation (leaking of the valve)
- Facial (and Head) Features (Figure 7)
- Widely spaced eyes (hypertelorism)
- Flat cheekbones (malar hypoplasia)
- Early fusion of joints between the skull bones (craniosynostosis)
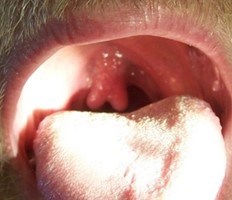
- Split (bifid) or broad uvula (the little piece of flesh that hangs down in the back of the throat) (Figure 8)
- An opening or split in the roof of the mouth (cleft palate)
- Small and recessed chin
- Eye Problems
- Nearsightedness (myopia)
- Eyes that don’t point in the same direction (strabismus)
- Eye muscle disorders
- Separation of the retina from the back of the eye (retinal detachment)
- Blue or grey sclera (bluish color of the white part of the eye) (Figure 7)
- Skeletal Symptoms
- Feet severely turned inward (clubfoot)
- Curving of the spine (scoliosis)
- Weak connections between the spinal bones of the neck and skull (cervical-spine instability)
- Sunken chest (pectus excavatum) or protruding chest (pectus carinatum)
- Contractures of the fingers (campylodactyly)
- Chiari malformation (abnormal findings on brain imaging)
- Skin Symptoms
- Abnormal scarring (widened)
- Easy bruising/bleeding
- Very soft skin
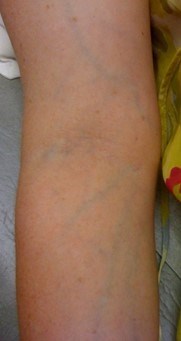
- Translucent skin (can easily see the veins under the skin) (Figure 9)
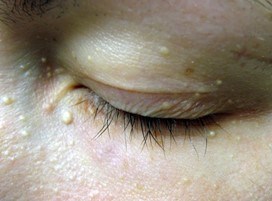
- Milia (small white dots) on the face (Figure 10)
- Other Symptoms
- Hernias
- Food or environmental allergies
- Inflammatory bowel disease
- Asthma
In Loeys-Dietz, there can be quite a bit of variability in the outward skin and skeletal features and problems with the heart and blood vessels, even among individuals affected from the same family with the same genetic mutation causing LDS.
- The features that help individuals and doctors consider the presence of LDS include:
- Aortic and branch vessel aneurysms: These are detected by medical imaging tests like echocardiogram, CT, and MRI scans. The most common aneurysm is at the beginning of the aorta (Figure 1, Figure 2) (the main blood vessel carrying blood from the heart out to the body). It is important to note that other parts of the aorta and other arteries throughout the body can also have aneurysms in LDS.
- Arterial tortuosity: Most often found in the head and neck and may be in the thoracic or abdominal aorta; can be observed on medical imaging tests. (Figures 3-5)
- Hypertelorism (Figure 7)
- Bifid uvula (Figure 8) or a cleft palate
It is important to note that not every individual with LDS has all four of these features. Some people with LDS may not even have external features (and thus are called non-syndromic). In addition, some individuals with LDS will have features that overlap with other more common conditions such as Marfan syndrome. These individuals may be initially thought to have Marfan syndrome until genetic testing confirms the mutated gene causing LDS instead. There are a few features that can specifically differentiate those with Marfan and those with LDS. People with Marfan syndrome do not typically have widely spaced eyes, a split uvula, or a cleft palate.
(from Narayanapillai J. Heart Views. Apr-Jun 2017;18(2):64-65. doi: 10.4103/1995-705X.208668. Copyright : © 2017 Heart Views)
(from Setty R, et al. Int J Surg Case Reports: 2018; 63: 113-117.)
What kind of testing is needed?
It is recommended that individuals be evaluated by doctors who are familiar with the features of connective tissue disorders like LDS. During the first visit, a detailed family and medical history is taken, and a complete physical exam will take place to look for the typical features in people with LDS. It is helpful to gather information before the visit about any family member who may have LDS, who had an aneurysm, or who had an aortic dissection (tear in the wall of the aorta).
Because one of the major features of LDS is an enlargement of the aorta, it is necessary for the doctor to get an image of one’s heart with an echocardiogram (Figure 11) (imaging using soundwaves). An echocardiogram is recommended at least every year depending on the size and rate of growth of the aorta.
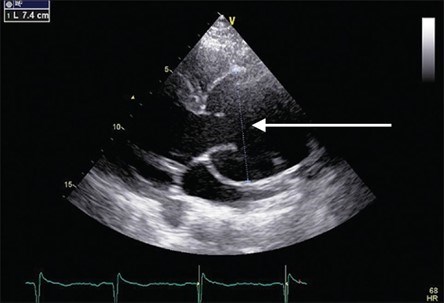
(from Narayanapillai J. Heart Views. Apr-Jun 2017;18(2):64-65. doi: 10.4103/1995-705X.208668. Copyright: © 2017 Heart Views)
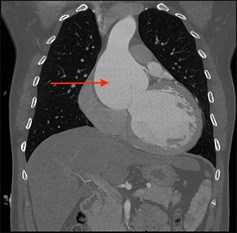
Recommendations are also made for additional imaging of the arteries throughout the body from the head to the hips. This typically involves ordering a CTA (computed tomography angiogram which uses x-rays to image- Figure 2) or an MRA (magnetic resonance angiogram which uses a magnetic field to generate images). The frequency of MRA or CTA imaging of the entire aorta and other blood vessels depends on how much the other vessels are involved, but it may be at least every two years.
Three-dimensional (3D) reconstruction of the CTA or MRA imaging is recommended to check for the presence of arterial tortuosity in the neck (Figure 3). The cervical (neck) spine may be abnormal in LDS, so x-rays of the cervical spine are recommended to determine if there is a problem. If they find significant abnormalities, it is recommended that individuals consult with an orthopaedist (bone doctor) or neurosurgeon (brain surgeon).
Genetic testing for a mutation within one of the six genes (TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3 and SMAD2) causing LDS is available and should be performed when someone has features of LDS. When LDS is found in an individual, evaluation of other family members is necessary. If a gene mutation causing LDS is found in a child, it is typically recommended to test the parents for the same mutation. This is to see if there is a risk of having another child with LDS. If a person has LDS, each child they have has a 50% chance of also getting LDS. Additionally, genetic testing of the children and siblings of an adult diagnosed with LDS is recommended.
How is LDS treated?
While LDS is a disorder that cannot be cured, survival can be greatly improved with proper treatment and management. The severity of this condition varies greatly from person to person. For most people, management will consist of daily heart medications and some physical activity restrictions. Routine doctor appointments and imaging tests are required. These may be yearly or more frequent, depending on the size of the aorta and the severity of other symptoms.
Medications: In an effort to slow the rate of aortic growth, individuals with LDS will be given some medications. The medications typically used are often the same ones used to treat people with high blood pressure:
- Angiotensin receptor blockers or ARBs (such as losartan or irbesartan)
- Beta-blockers (such as atenolol or metoprolol), sometimes in combination with ARBs
It is recommended that individuals with LDS remain on their medications even if they have surgery to fix an aneurysm. The US Food and Drug Administration (FDA) also recently warned that people with LDS and other aortic aneurysm conditions should avoid commonly used antibiotics called fluoroquinolones (includes Avelox, Cipro, Factive, Levaquin, and Ofloxacin). The reason for this recommendation is that recent medical studies have shown that people who have taken a fluoroquinolone are twice as likely to have an aortic aneurysm or dissection than those who have not taken one of these drugs.
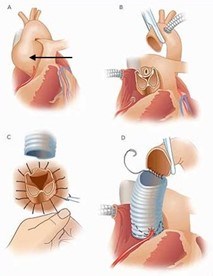
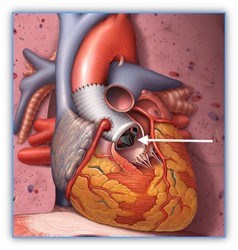
Surgery: Depending on the size or rate of growth of an aneurysm or artery (usually the aorta), surgery may be recommended to reduce the risk of dissection or vessel rupture. If someone has a family history of early aortic dissection, or if their LDS is caused by certain mutations, their care provider may recommend surgery earlier than usual and at an aortic diameter smaller than is recommended for people with Marfan syndrome. In general, preventative aortic root replacement for LDS due to mutations in TGFBR1 and 2 is typically recommended when the aortic root diameter reaches 4.0 to 4.5 cm, depending upon the gene mutated, age, sex, family history, rate of aortic growth, and other features of the condition. For instance, women with TGFBR2 mutations and more severe features of LDS may be at higher risk and surgery at a smaller aortic diameter may be recommended. There is not a lot of information about timing of surgery in the other types of LDS, and a shared decision with a cardiologist, geneticist, and cardiac surgeon is required. Aortic root replacement is the most common aortic surgery for people with LDS, and it is highly successful in centers that have surgeons experienced with their surgery in people with connective tissue conditions. It is likely more successful when the surgery is performed electively (for prevention) as opposed to needing the surgery in the case of an emergency (such as with an aortic dissection). In most cases, the valve separating the heart and the aorta can be left in place during an aortic root replacement (so called valve sparing root replacement, Figure 12). This means warfarin (a blood thinner) is not required, though it is used for people who have a mechanical aortic valve. In some cases, the valve will have to be replaced (composite valve graft, Figure 13) and a blood thinner is required.
Are there limitations on exercise?
Most people with LDS can and should be physically active; however, they should not exercise to the point of becoming exhausted. They should focus on low-intensity (aerobic) and low-impact activities. Nearly every physical activity can be done at different intensity levels. Individuals need to discuss physical activities and activity levels with their physician before beginning any exercise program, and especially as they age.
- Competitive and contact sports increase the risk of injury. Doctors worry about:
- Increased heart rate and blood pressure can put added stress on the aorta
- Stress on the bones and joints can lead to added pain and joint dislocations
- Bruising and internal bleeding due to certain medications (such as blood thinners)
- There are several things to consider when deciding if a sport or activity is safe for someone with LDS:
- Favor non-competitive activity
- Perform activities at a non-strenuous pace
- Choose an enjoyable activity that can be performed 4–5 times per week for 30 minutes at a time
- Stay at an aerobic level of work (not breathing hard and could easily carry on a conversation during the activity)
- Wear protective gear
- Consider traditional yoga for relaxation and stress reduction
- Individuals should not test their limits
- When choosing an activity, avoid the three C’s: competition, collision, and contact. Exercises to avoid include:
- Intense isometric exercises (where specific muscles are worked without moving, such as weightlifting, pull-ups, push-ups, planking or wall sits, or those leading to muscle straining).
- Exercise to the point of exhaustion (breathless, unable to speak)
Overall, a heart-healthy lifestyle should be followed. Smoking and illicit drug use should be avoided, and traditional cardiovascular risk factors such as high cholesterol or high blood pressure should be aggressively managed.
How does LDS affect one’s ability to start a family?
Women with LDS have an increased risk of aortic dissection during pregnancy and the time right after delivery. For this reason, a consultation with a cardiologist and high-risk obstetrical or maternal-fetal medicine specialist is recommended before considering pregnancy. If there is significant aortic enlargement, the risk of aortic dissection is higher. If an aortic aneurysm is present, a cardiologist may recommend against becoming pregnant. Effective birth control methods should be discussed to avoid an unplanned pregnancy. For women who carry the gene changes found in LDS but who have less of the common features, the risk of an aortic dissection related to pregnancy may be lower.
In addition, the associated risks of pregnancy may also vary depending on the LDS subtype. Having had an aortic aneurysm surgery in the past doesn’t necessarily reduce the risks of pregnancy in LDS. There are also reports of uterine rupture in LDS. More frequent aortic imaging follow-up during pregnancy and immediately after delivery is necessary. Medications may need to be adjusted before pregnancy because coumadin and angiotensin receptor blocker medications (ARBs, such as losartan) may harm the developing baby.
In addition to careful assessment of the risk for women contemplating pregnancies, couples in whom the underlying genetic defect is known may consider prenatal or pre-implantation diagnostics.
- Prenatal testing
- Prenatal testing is the technique in which early in the pregnancy (11-12w) the presence of the genetic defect is checked by means of a so-called villous sampling in which a piece of placenta (which is of fetal origin) is punctured away. If the result is abnormal, the pregnancy will be interrupted. With this technique, pregnancy occurs naturally but there is a 50% chance that the pregnancy must be interrupted.
- Pre-gestational diagnostic
- PGD is a technique that was developed more recently and where embryos are developed via an in vitro fertilization procedure. For this, oocytes are removed from the ovaries after stimulation and are fertilized with sperm in a test tube. In an early stage of embryo development, one cell is subsequently removed for testing for the presence of the genetic defect. Only non-affected embryos are replaced. With this technique, the pregnancy is artificially established.
In both cases, knowledge of the pathogenic variant is an absolute requirement. Dedicated pre-conceptual counseling is also necessary.
What are the signs of an acute aortic event? What should be done if one occurs?
Aortic dissection (a tear in the wall of the aorta) is very dangerous and must be treated as soon as possible.
We think there are probably 5,000-10,000 dissections a year in the United States. However, it is likely that many dissections are reported as a “heart attack” or “sudden death,” when the cause is actually a dissection.
Individuals with Loeys-Dietz syndrome are at much greater risk for an aortic dissection than the general population. There are differences in risks for aortic dissection based on the genes causing LDS. It is important to know the signs of an aortic dissection and what to do.
Signs and symptoms of acute aortic dissection may include:
- Sudden onset, severe or sharp chest, back, neck/head, or abdominal pain
- Pain described as “ripping” or “tearing”
- Pain radiating (traveling) from the chest to the back, head and neck, abdomen, or legs
- Chest pain associated with fainting (syncope)
- Acute shortness of breath
- Acute stroke symptoms or inability to use an arm or leg
- Unexplained pain and the feeling of “something is very wrong” or a “sense of doom”
Chest pain can have many causes, but for those with LDS, it is very important to check for aortic dissection. If symptoms are present that could be due to an aortic dissection, calling 911 and getting to an emergency room right away is necessary. A known LDS diagnosis will help if an individual has unexplained chest, back, or abdominal pain. Other causes for acute chest pain or shortness of breath in individuals with LDS include a spontaneous collapsed lung (pneumothorax).
Since emergency rooms may be very busy and most likely don’t know a lot about LDS, individuals with LDS and their family members should plan to advocate for themselves if they have these symptoms and end up in an ER. Individuals with LDS should tell the doctors and nurses at the hospital what they know about their condition. By telling their doctors, nurses, and emergency personnel that they have LDS and are concerned about the symptoms related to an aortic dissection, affected individuals can be more quickly evaluated for the condition.
A medical alert bracelet can alert emergency personnel right away that an individual is at risk for a life-threatening dissection.
Where can I learn more about Loeys-Dietz syndrome?
The Loeys-Dietz Syndrome Foundation at http://www.loeysdietz.org/
The Marfan Foundation — related disorders page at https://www.marfan.org/loeys-dietz
Genetic Aortic Disorders Association of Canada http://www.gadacanada.ca/loeys-dietz-syndrome
The National Center for Biotechnology Information https://www.ncbi.nlm.nih.gov/books/NBK1133/